- +1 858 909 0079
- +1 858 909 0057
- [email protected]
- +1 858 909 0079
- [email protected]

Products
Specification
Composition
Magnetic beads grafted with hydroxyapatite group.
Beads Size
~ 1.0 μm diameter
Magnetization
~45 EMU/g
Type of Magnetization
Superparamagnetic
Effective Density
2.0 g/ml
Stability
Most organic solvents
Formulation
Lyophilized Powder
Binding Capacity
>25μg BSA/ mg of Beads
Storage
Store at 4°C upon receipt.
Hydroxyapatite, also known as hydroxylapatite (HA), is a naturally occurring mineral with a chemical formula of Ca5(PO4)3(OH)2. It has a composition and morphology like that of human hard tissues. One of the unique properties of hydroxyapatite is that its surface can have a mixed charge, which can be either positively or negatively charged depending on the pH of the surrounding environment. This article will discuss the use of hydroxyapatite-based chromatography columns for the purification of proteins, enzymes, DNA, and RNA in the biotechnology and pharmaceutical industries.
BcMag™ Hydroxyapatite Magnetic Beads, the magnetic particles boasting a uniform coating of hydroxyapatite functional groups on their surface (as illustriously portrayed in Figure 1), are a highly sought-after commodity in the realm of modern scientific research. These beads, with their unparalleled prowess, can expediently process an astounding number of 96 samples within a mere 20 minutes, delivering yields that are nothing short of spectacular. Not only do they boast of their sheer processing speed, but they are also highly efficient in purifying antibodies, nucleic acids, viruses, and other large molecules from complex biological samples.
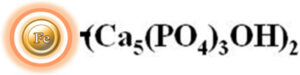
●
Fast and straightforward – Magnetic beads-based format eliminates columns or filters or a laborious repeat of pipetting or centrifugation.
●
Convenient and expandable – Magnetic format enables high-throughput processing of multiple samples in parallel with many different automated liquid handling systems
●
Robust – Magnetic resins do not crack or run dry.
●
Low bed volume – Working with small magnetic bead volumes allows for minimal buffer volumes, resulting in concentrated elution fractions.
●
Chemical and thermal stability – A wide range of chemical compatibilities (aqueous and inorganic solvents), heat stability (autoclavable), and pH tolerance (pH >5.5) let hydroxyapatite be utilized below settings that improve nucleic acid and protein binding.
Note:
The following protocol is an example of purifying proteins with BcMag™ hydroxyapatite-modified magnetic beads. Users may use alternative binding, washing, or elution buffers and are encouraged to determine the optimal working conditions based on the protocol and suggestions described in the Note sections. We recommend optimizing the amounts of beads used for each application. Adjust elution volumes to avoid unnecessary sample dilution.
Materials Required
●
Magnetic Rack (for manual operation)
Based on sample volume, the user can choose one of the following Magnetic Racks:
– BcMag™ Magnetic Rack-2 for holding two individual 1.5 ml centrifuge tubes (Cat. No. MS-01);
– BcMag™ Magnetic Rack-6 for holding six individual 1.5 ml centrifuge tubes (Cat. No. MS-02);
– BcMag™ Magnetic Rack-24 for holding twenty-four individual 1.5-2.0 ml centrifuge tubes (Cat. No. MS-03);
– BcMag™ Magnetic Rack-50 for holding one 50 ml centrifuge tube, one 15 ml centrifuge tube, and four individual 1.5 ml centrifuge tubes (Cat. No. MS-04);
– BcMag™ Magnetic Rack-96 for holding a 96 ELISA plate or PCR plate (Cat. No. MS-05).
For larger scale purification, Ceramic Magnets Block for large scale purification (6 in x 4 in x 1 in block ferrite magnet, Applied Magnets, Cat. No. CERAMIC-B8).
●
Corning 430825 cell culture flask for large-scale purification (Cole-Parmer, Cat. No. EW-01936-22)
●
Mini BlotBoy 3D Rocker, fixed speed, small 10″ x 7.5″ platform w/ flat mat (Benchmark Scientific, Inc. Cat. No. B3D1008) or compatible
A. Protein purification
Note:
●
Monobasic sodium phosphate monohydrate and dibasic sodium phosphate 7-hydrate are recommended for Precondition Buffer and Binding/Wash buffer preparations. Avoid anhydrous sodium phosphate because these salts contain pyrophosphate that prevents the binding of some macromolecules.
●
Avoid High concentrations of salt or chelating agents such as EDTA because they will prevent proteins from binding to the magnetic beads.
●
Calcium chloride may be added to the phosphate buffer to increase the binding efficiency of acidic proteins. Concentrations of calcium chloride in phosphate buffers: 0.3 mM calcium chloride for 10 mM phosphate buffers: 0.01 mM calcium chloride for 300 mM phosphate, and 0.0075 mM calcium chloride for 400 mM phosphate.
Buffers
●
Precondition Buffer: 200 mM Sodium phosphate, pH 9-10
●
Binding/Wash Buffer: 10 mM Sodium phosphate, pH 6.8, 0.3 mM calcium chloride
●
Elution Buffer: Gradient of increasing concentration of 10-600 mM potassium phosphate, pH 7.0
a.
Sample preparation
1.
Dialyse the protein sample against 50 volumes of Binding buffer.
b.
1.
Shake the bottle and completely resuspend the Magnetic Beads.
2.
Transfer 40μl magnetic beads (2 mg) to a centrifuge tube. Place the tube on the magnetic separator for 1-3 minutes. Remove the supernatant while the tube remains on the separator.
3.
Remove the tube and resuspend the beads thoroughly with 200μl Precondition Buffer. Leave the tube at room temperature for 2-3 minutes. Place the tube on the magnetic separator for 1-3 minutes. Remove the supernatant while the tube remains on the separator.
4.
Repeat step 3 once.
5.
Remove the tube from the separator and resuspend the beads thoroughly with a 200μl Binding/Wash Buffer. Place the tube on a magnetic separator for 1-3 minutes. Remove the supernatant while the tube remains on the separator.
6.
Repeat step 5 once.
7.
Resuspend the beads thoroughly with 100 μl Binding/Wash Buffer.
1.
Add your desired sample containing ~40µg protein to the tube containing the washed beads from step B.7.
2.
Mix beads well with a pipette and leaves them at room temperature for 2-3 minutes. Place the tube on a magnetic separator for 1-3 minutes. Remove the supernatant while the tube remains on the separator.
3.
Place the tube on the magnetic separator for 1-3 minutes. Remove the supernatant while the tube remains on the separator. Remove the tube from the separator and wash the beads with a 200µl Binding/Wash Buffer.
4.
Repeat step 3 for three times.
Note:
Protein can be eluted with a gradient of increasing phosphate buffer concentration (10-600 mM) and pH gradient (5.5 or higher, up to the stability limit of the sample protein), or NaCl for basic proteins, but not for acidic proteins.
1.
Remove the tube from the separator. Resuspend the beads with 10-20 µl Elution Buffer and leave them at room temperature for 3 minutes.
2.
Place the tube on the magnetic separator for 1-3 minutes and transfer the supernatant containing the eluted protein to a new tube.
B. DNA purification
Buffer
Note:
Monobasic sodium phosphate monohydrate and dibasic sodium phosphate 7-hydrate are recommended for all buffer preparations. Avoid anhydrous sodium phosphate because these salts contain pyrophosphate that prevents the binding of some macromolecules.
●
Sample lysis Buffer: 8M Guanidine hydrochloride
●
Binding/Washing Buffer: 100 mM phosphate buffer pH 7, 4 M guanidine hydrochloride
●
Elution Buffer: 0.5M phosphate buffer pH 7 (4M guanidine hydrochloride -optional)
●
Dilution Buffer: 0.2 M phosphate buffer pH 7
a.
Sample Preparation:
Note:
Sample pre-treatment is a critical step for successfully purifying high-quality genomic DNA. Different biological samples require different methods to release their genomic DNA from cells. Many cultured cells can be efficiently homogenized in lysis buffer by vortex, while animal/plant tissues, yeasts, and bacteria need a more powerful lysis process. Suggested methods for preparing different samples are listed in the following table.
Sample
Sample
Liquid Nitrogen
Frozen Grinding
Homogenize
Lysozyme
Lyticase, Zymolase
Sonication
Glass Bead Grinding
Soft Tissue
+
+
Hard Tissue
+
+
+
Plant Tissue
+
+
Fungi
+
+
Yeast
+
+
Bacterium
+
+
1.
Dissolve 100 mg pre-treated biomass into 1ml of sample lysis buffer. To promote lysis, incubate at 55 °C for 1 hour to overnight (depending on the sample) on a shaker-incubator
2.
Centrifuge at 13,000 rpm for 3 ~ 5 minutes at room temperature to remove cell debris. Transfer the supernatant to a new tube and adjust the concentration of the supernatant to 4 M guanidine HCl with 2x Dilution buffer.
b.
1.
Shake the bottle to resuspend the Magnetic Beads completely.
2.
Transfer 20μl-30μl magnetic beads (50 mg/ml) to a centrifuge tube. Place the tube on the magnetic separator for 1-3 minutes. Remove the supernatant while the tube remains on the separator.
3.
Remove the tube and resuspend the beads thoroughly with 200μl Binding/Washing. Leave the tube at room temperature for 2-3 minutes. Place the tube on the magnetic separator for 1-3 minutes. Remove the supernatant while the tube remains on the separator.
4.
Repeat step 3 three times.
5.
Resuspend the beads thoroughly with 100 μl Binding/Wash Buffer.
1.
Mix the sample with the prepared beads and incubate at room temperature for 10-15 minutes with gentle rotation.
2.
Place the tube on a magnetic separator for 1-3 minutes. Remove the supernatant while the tube remains on the separator.
3.
Remove the tube from the separator and wash the beads with a 200µl Binding/Wash Buffer. Place the tube on the magnetic separator for 1-3 minutes. Remove the supernatant while the tube remains on the separator.
4.
Repeat step 3 for three times.
1.
Remove the tube from the separator, resuspend the beads with 10-50 µl Elution Buffer and leave them at room temperature for 3 minutes.
2.
Place the tube on the magnetic separator for 1-3 minutes and transfer the supernatant containing the eluted DNA to a new tube.
1.
Add an equal volume of d2H2O to the eluted DNA solution
2.
Add 0.1 volume 5M ammonium acetate and 2.5 volumes of 100 % EtOH to precipitate the DNA for at least 15 minutes (1 hour or longer is preferred) at -20 °C
3.
Centrifuge for 15 mins at 13,000 rpm. Discard the supernatant. Add 2 ml of ice-cold 70 % ethanol to the pellet and flick/agitate the tube to resuspend the pellet.
4.
Centrifuge for 15 mins at 13,000 rpm.
5.
1.
Gagnon P. Monoclonal antibody purification with hydroxyapatite. N Biotechnol. 2009 Jun;25(5):287-93.
2.
Gagnon P, Beam K. Antibody aggregate removal by hydroxyapatite chromatography. Curr Pharm Biotechnol. 2009 Jun;10(4):440-6.
3.
Wang Y, Carta G. Competitive binding of monoclonal antibody monomer-dimer mixtures on ceramic hydroxyapatite. J Chromatogr A. 2019 Feb 22;1587:136-145.
4.
Cummings LJ, Frost RG, Snyder MA. Monoclonal antibody purification by ceramic hydroxyapatite chromatography. Methods Mol Biol. 2014;1131:241-51.
5.
Cummings LJ, Snyder MA, Brisack K. Protein chromatography on hydroxyapatite columns. Methods Enzymol. 2009;463:387-404.
6.
Hilbrig F, Freitag R. Isolation and purification of recombinant proteins, antibodies and plasmid DNA with hydroxyapatite chromatography. Biotechnol J. 2012 Jan;7(1):90-102.
7.
Broadhurst AV. Hydroxylapatite chromatography. Curr Protoc Protein Sci. 2001 May;Chapter 8:Unit8.6.
8.
Niimi M, Masuda T, Kaihatsu K, Kato N, Nakamura S, Nakaya T, Arai F. Virus purification and enrichment by hydroxyapatite chromatography on a chip. Sens Actuators B Chem. 2014 Oct 1;201:185-190.
9.
Duffy E, Florek J, Colon S, Gerdon AE. Selected DNA aptamers as hydroxyapatite affinity reagents. Anal Chim Acta. 2020 May 8;1110:115-121.
10.
Brundin M, Figdor D, Sundqvist G, Sjögren U. DNA binding to hydroxyapatite: a potential mechanism for preservation of microbial DNA. J Endod. 2013 Feb;39(2):211-6.
Get the Latest News and Updates by Email
6393 Nancy Ridge Dr. Suite A
San Diego, CA 92121 USA
Fax: +1-858-909-0057
Get the Latest News and Updates by Email
© 2023 Bioclone Inc. All Rights Reserved.
Magnetic Beads Make Things Simple